摘 要:目的:研究绿色荧光蛋白(Greed Fluorescent Protein,GFP)基因的基因克隆及在大肠杆菌中的表达。方法:通过分别将DH-5α (pEGFP-N3)和DH-5α(pET-28a)提取质粒、酶切并连接形成重组质粒pET-28a-GFP,将重组质粒导入E.coli DH-5α感受态细胞中进行转化,通过限制性核酸内切酶Not I与Bam H1和PCR对所建质粒进行分析鉴定后, 通过转化的方法把含绿色荧光蛋白(GFP)外源基因转入大肠杆菌体BL-21内进行表达,再用IPTG诱导GFP基因表达,可以看到显现绿色,判断GFP基因在大肠杆菌中成功表达。 结果:结果显示构建的重组质粒pET-28a-GFP在E.coli中成功表达。
关键词:绿色荧光蛋白;质粒重组;原核表达;诱导表达
中图分类号:Q53
Studies On Cloning and Expression of Green Fluorescent Protein gene
Abstract: Objective:Studies indicated that the cloning and expression of the GFP gene in the E.coli. Methods: Extract the plasmid of the DH-5α(pEGFP-N3) and DH-5α (pET-28a). Then cutting by enzyme and connecting the two plasmids to form pET-28a-GFP recombined plasmid. The recombinant plasmid confirmed by restriction enzyme and PCR transfected into E.coli DH-5α to ensure the expression of green fluorescent protein. Guiding the recombined plasmid, which contains exogenous genes of GFP into E.coli for expression, through transformative method. The expression of GFP gene can be induced by the IPTG and then we can see green. Results: The results suggest that pET-28a-GFP recombined plasmid has successfully expressed in E.coli.
Keywords: Ged Fluorescent Protein; Recombined Plasmid; Prokaryote Expression; Induced Expression
绿色荧光蛋白的基因克隆和表达的研究
引言
随着分子生物学和基因工程技术的迅速发展和广泛应用, 人们根据自己的意愿有目的、有计划、有根据、有预见地将外源基因导入动物细胞内, 使外源基因进行表达、阐明基因表达的调控机理或者通过与染色体基因组进行稳定整合,将生物性状传递给子代动物的研究方兴未艾
[1]。
基因标记技术是近年来发展起来的分子生物学技术。荧光蛋白基因在标记基因方面由于具有独特的优点而引起了科学家的广泛关注,现已被普遍应用到分子生物学研究的各个方面。荧光蛋白是海洋生物体内的一类发光蛋白,分为绿色荧光蛋白、蓝色荧光蛋白、黄色荧光蛋白和红色荧光蛋白
[2]。
2008 年10 月8 日,瑞典**科学院把今年的诺贝尔化学奖授予绿色荧光蛋白的发现者和推广者。他们分别为日本科学家下村修(Osamu Shimomura)、美G科学家马丁·查尔菲(Martin Chalfie)和钱永健(Roger Tsien)
[3]。1962 年,下村修等分离纯化了水母中发光蛋白水母素,并发现一种绿色的荧光蛋白。1974 年,他们分离得到了这个蛋白,当时称绿色蛋白,以后称绿色荧光蛋白(GFP)
[4] 。1994 年,查尔菲等**在大肠杆菌细胞中表达了能发射绿色荧光的GFP,开创了GFP 研究与应用之先河
[5]。
绿色荧光蛋白( green fluorescent p rotein, GFP)是238 个氨基酸组成的单体蛋白,其分子量为27 kDa。GFP 作为一种新的报告基因,与以往lacZ、CAT 等报告基因相比,有很多无可比拟的优越性: GFP 不具有种属依赖性,在多种原核和真核生物细胞中都表达;荧光强度高,稳定性高;不需要反应底物与其他辅助因子,受蓝光激发产生绿色荧光,尤其适用于体内的即时检测;另外GFP 分子量小,易于融合,适用于多种转化方式,对受体无毒害,安全可靠;并且通过替换一些特殊氨基酸,可以使之产生不同颜色的光,从而适应不同的研究需要。正是由于GFP 检测具有高灵敏度,操作简单,无需使用同位素等优点,近年来广泛用于基因的表达与调控、蛋白质的定位、转移以及相互作用、信号传递、转染与转化,以及细胞的分离与纯化等研究*域
[ 6~7] 。采用GFP 作为标记基因,可直接收集转化细胞供实验,缩短了筛选时间、减少对细胞活性的影响并可作为活体标记,为研究发育的基因调控和分子机制提供了一种简洁有效的手段
[ 8、9 ] 。采用基因工程手段生产GFP 标记的方法,可建立一种简便、快速的免疫诊断技术
[10] 。
质粒转化进入大肠杆菌(
Escherichia coli)感受态细胞是分子克隆的关键步骤
[11],是基因克隆以及DNA文库构建等研究中频繁使用的一项重要的常规操作。一般实验室没有电穿孔技术所需要的特殊仪器,而利用氯化钙法将质粒重组入大肠杆菌细胞,操作方便、应用广泛.目前, 感受态细胞的制备主要采用CaCl
2 法, 该方法操作简单、容易掌握、重复性好、转化率高, 可广泛应用于一般的实验室。其原理是Ca
2+ 破坏细胞膜上的脂质阵列,并与膜上多聚羟基丁酸化合物、多聚无机磷酸形成复合物以利于外源DNA 的渗入
[12]#p#分页标题#e#。
PCR 技术是Kary Mullis 在1985 年建立起来的在细胞体外合成DNA 的一种方法。依DNA 半保留复制原理,利用DNA聚合酶依赖于DNA 模板的特性,在附加的两个引物与模板杂交之后,按碱基配对原则经酶促反应合成DNA 片段,包括模板变性、引物退火及用DNA聚合酶延伸两个引物之间DNA 的一定次数的重复循环,使包括在两个引物5'端限定的特异性片段形成指数式积累。整个过程操作简单,可在短时间内在小管中获得大量的特意的DNA 拷贝,这一特点使PCR 技术很快被应用到了分子生物研究的广大*域
[13]。
大肠杆菌是**个用于重组蛋白生产的宿主菌,它不仅具有遗传背景清楚、培养操作简单、转化和转导效率高、生长繁殖快、成本低廉、可以快速大规模地生产目的蛋白等优点
[14]。而且其表达外源基因产物的水平远高于其它基因表达系统,表达的目的蛋白量甚**能超过细菌中蛋白量的30 %,因此大肠杆菌是目前应用**广泛的蛋白质表达系统
[15]。大肠杆菌表达系统是目前**常用的外源蛋白表达系统,大肠杆菌由于遗传背景清楚、易操作,以及有大量可供选择的克隆或表达载体,使之成为人们克隆表达外源基因的主要菌株
[16]。
随着人们对原核和真核生物基因调控的了解,通过综合控制基因转录、翻译、蛋白质稳定性及向胞外分泌等诸多方面的因素,构建出了多种具有不同特点的表达载体和工程菌株,以满足表达不同性质、要求的目的基因的需要。在原核细胞中表达蛋白质的载体常用启动子有T7启动子、Trp启动子-色氨酸启动子、Tac启动子、T7噬菌体中基因10 的启动子及Lac启动子等。Lac启动子受分解代谢系统活化蛋白和cAMP的正调控和阻遏物的负调控,当加入乳糖或某些类似物IPTG可与阻遏蛋白形成复合物,使阻遏蛋白构型改变,阻遏蛋白不再能与操纵基因结合,从而使结构基因表达。根据原核生物基因表达特点选着载体进行目的基因克隆,然后转入相应的菌种中表达目的蛋白质
[17]。
研究绿色荧光蛋白在大肠杆菌体内的基因克隆和表达。通过质粒重组形成所需要的重组质粒pET-28a-GFP,将重组质粒导入大肠杆菌体内,通过酶切、PCR及用IPTG诱导检测是否在大肠杆菌体内诱导表达成功。根据电泳结果及荧光现象得出结论,重组质粒在大肠杆菌体内成功诱导表达。
1 材料与方法
1.1 材料
1.1.1 实验材料
克隆菌
E.coli DH-**、表达菌
BL-21为本实验室收藏菌种,质粒 pET-28a 和 pEGFP-N3,引物,限制性内切酶 Bam H1、 Not Ⅰ购自大连宝生物工程有限公司。
1.1.2 仪器设备
SIM-f140 SANYO Ice Maker(made in Japan)
Eppendof离心机5430R(Made in Germany)
电泳仪 (DYY-12型,北京市六一仪器厂)
电子天平 (AB204-N 型,Mettler-Toleda Group)
台式离心机 (TGL-16C型,上海安亭科学仪器厂)
控温磁力搅拌器 (HJ-3型,江苏金坛市金南仪器厂)
调温电热套 (KDM型,武汉精华科教仪器有限公司)
ph计(雷磁PHS-3C型,上海精密科学仪器有限公司)
冰箱 (BCD-208K/A NCJN型,青岛海尔股份有限公司)
台式冷冻恒温振荡器 (THZ-C-1型,太仓市实验设备厂)
手提紫外灯(WD-9403E型,北京市丰台区造甲街128号)
生物洁净工作台 (BCM-1000型,苏州净化设备有限公司)
电热恒温水温箱(SHW 21-420型,湖北省黄石市恒丰医疗器械有限公司)
琼脂糖凝胶电泳电泳装置、凝胶成像分析系统、酒精灯、培养皿、
移液枪、枪头 、接种环 、酒精棉球 、灭菌枪头 、Parafilm膜、离心管
1.1.3 试剂及溶液
|
50×TAE电泳缓冲母液(50 mL) |
|
|
Tris-碱 |
12.1 g |
|
冰醋酸 |
2.85 mL |
|
0.5 mol/L EDTA (pH 8.0) |
5 ml |
|
6×DNA电泳缓冲液 |
|
|
溴酚蓝 |
0.25 % |
|
蔗糖水溶液 |
40%(W/V) |
|
液体LB培养基(pH7.0) |
|
|
胰蛋白胨 |
10 g/L |
|
酵母提取物 |
5 g/L |
|
NaCl |
10 g/L |
|
NaOH(1 mol/L) |
1 mL/L |
分装后于121 ℃ 高压灭菌20 min。(LB固体培养基是在液体LB中加琼脂粉**1 %);#p#分页标题#e#
|
溶液Ⅰ 50 mL |
|
|
葡萄糖 |
50 mmol/L |
|
Tris-Cl (pH 8.0) |
25 mmol/L |
|
EDTA (pH 8.0) |
10 mmol/L |
121℃高压灭菌 15 min后置于0~4℃贮存;
|
溶液Ⅱ 100 mL |
|
|
NaOH |
0.2 mol/L |
|
SDS |
1% (W/V) |
用时由母液2 mol/L NaOH、10%(W/V) SDS稀释现配;
|
溶液Ⅲ 100 mL |
|
|
KOAc (5 mol/L) |
60 mL |
|
冰乙酸 |
11.5 mL |
|
H2O |
28.5 mL |
121 ℃高压灭菌 15 min后置于0~4 ℃贮存;
氯仿;
琼脂糖;
灭菌的去离子水;
10×酶切缓冲液;
TaqDNA聚合酶;
TaqDNA聚合酶缓冲液;
灭菌的0.1 mol/L CaCl
2
标准相对分子质量的DNA;
溴乙锭(EB)储存液 0.5 µg/mL;
LB/Kan(50 µg/mL)固体培养基;
IPTG配成浓度为400 mM水溶液,-20 ℃保存备用;
卡那霉素(Kan)配成浓度为100 ug/mL水溶液,-20 ℃保存备用;
70%乙醇:用新开装的乙醇和灭菌无离子水配成,放在0~4 ℃;
无水乙醇(分析纯,武汉市中天化工有限公司)放在0~4 ℃;
RNase 母液:将RNase 溶于10 mmol/L Tris·Cl(pH 7.5)、15 mmol/L NaCl 配成的试剂中,配成10 mg/mL的溶液,在100 ℃加热15 min,使可能溶有的DNase失活,然后缓慢冷却**室温,分装成小份保存于-20 ℃;
1.2 方法
1.2.1 重组质粒的构建
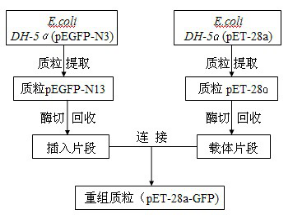
图1重组质粒pET-28a-GFP的构建流程
Fig.1 The formed technological process of pET-28a-GFP recombined plasmid
1.2.2 DH-5α 和BL-21感受态细胞的制备
1) 将0~4 ℃保存的
DH-5α 和
BL-21菌种分别接种在LB液体培养基中37 ℃下250 r/min过夜培养16 h 。
2) 将分别接种过夜菌:LB按1:50的比例接种于2 mL的LB液体培养基中,37 ℃活化培养2~3 h**OD=0.3~0.5 。
3) 取1.5 mL菌液转入EP管中,置于冰上10 min, 然后于4 ℃下5000 r/min离心5 min。弃上清液,沉淀加入0.1 mL预冷的0.1 mol/L CaCl
2缓和悬菌。冰上放置15-30 min后,4 ℃下5000 r/min离心10 min。
4) 弃上清液,沉淀用0.1 mL预冷的0.1 mol/L CaCl
2(含15%甘油)缓和悬菌,放在-20 ℃冰箱内保存。
1.2.3 感受态细胞的转化
1) 取制备好的感受态细胞100 μl,冰上解冻,均匀悬浮。
2) 加入2 μl酶连产物,轻轻混匀,冰上静置10-30 min。
3) 42 ℃水浴中热击70 sec后,冰上放置2 min。
4) 加入200 μl LB液体培养基,37 ℃,50-100 rpm振荡培养1 h。
5) 取200 μl悬浮细胞涂布在含合适抗生素的LB固体培养基上,用涂布器均匀涂布,平皿正放静置1-2 h后,封口膜封好平皿, 37 ℃培养倒置12-16 h。
1.2.4 碱法提质粒
1) 感受态细胞经转化培养后,平皿内有但菌落长出。
2) 用灭菌吸头挑取单菌落,浸没于2 mL含有抗生素的LB液体培养基中,37 ℃, 180 r/min振荡培养过夜。
3) 取1.5 ml 培养物倒入微量离心管中,用微量离心机于4 ℃、11000 r/min 离心1分钟,吸弃上清。
4) 向离心管中加入150 ml用冰预冷的溶液Ⅰ,用微量移液器吹打重悬沉淀。
5) 加入200 ml新配制的溶液 Ⅱ,盖紧管口,快速颠倒离心管5次,冰上放置5分钟。
6) 加入150 ml用冰预冷的溶液 Ⅲ,轻轻混匀,冰上放置3~5分钟。
7) 用微量离心机于4 ℃、11000 r/min离心5分钟,吸取上清转移到另一离心管。
8) 加等体积氯仿400 ml抽提,振荡混匀,用微量离心机于4 ℃、11000 rpm离心2分钟,将上清转移到另一离心管中。
9) 吸取上清液300 ml,向上清中加入2倍体积的无水乙醇,混匀后,于室温放置2分钟;用微量离心机于4 ℃、11000 rpm离心5分钟,弃上清。
10) 用70%乙醇洗涤2次,再次4 ℃、11000 rpm离心5分钟,沉淀于空气中干燥。向已干燥的离心管内加20 ml超纯水溶解质粒DNA,加入2 ml 10 mg/ml RNA酶,37 ℃处理1小时后于-20 ℃贮存。#p#分页标题#e#
1.2.5 酶切鉴定
1) 取提取的质粒8 ml于另一微量离心管中,加入2 ml Bam H I和Not I的酶切混合液,轻弹管外混匀反应物。
2) 离心,使溶液聚集在管底部。
3) 37 ℃,酶切反应。
1.2.6 琼脂糖凝胶电泳
1) 称取0.8 g琼脂糖,放入到锥形瓶中,加入100 mL 1×TAE缓冲液,置微波炉或水浴加热**完全融化,冷却**60 ℃左右。
2) 轻缓倒入封好两端和加上梳子的电泳胶板中,静置冷却30分钟以上。
3) 将胶板除去封胶带,加电泳缓冲液**电泳槽中,加液量要使液面没过胶面1-1.5毫米,轻轻拔除梳子。
4) 吸取10 ml的质粒与2 ml的上样液混匀,吸取混合液将加入加样孔。
5) 接通电泳槽与电泳仪的电源,设置电泳数据U=100 V, I=30 mA。
6) 当溴酚蓝染料移动到距凝胶前沿1-2 cm处,停止电泳。
7) 在凝胶成像分析系统中观察结果。
1.2.7 PCR-聚合酶链式反应
1) 将PCR管放置在冰盒上,按如下表格加入PCR反应体系各成分
|
PCR反应体系 |
各成分的量 |
|
template |
0.1 ml |
|
P1(20 mM) |
0.2 ml |
|
P2(20 mM) |
0.2 ml |
|
dNTPs(10 mM) |
0.4 ml |
|
Taqs(5 U/ml) |
0.2 ml |
|
10×PCR buffer |
2 ml |
|
ddH2O |
16.9 ml |
|
Total |
20 ml |
2) 将PCR管放入PCR仪中,设定PCR仪的循环参数。预变性95 ℃ 3 min,变性95 ℃ 30 s,退火60-55 ℃ 30 s,延伸72 ℃ 1 min 10个循环,每个循环降0.5 ℃。变性95 ℃ 30 s,退火55 ℃ 30 s,延伸72 ℃ 1 min 20个循环,72 ℃延伸 2 min。
3) PCR扩增完毕后,取出PCP管放在冰盒上。
4) 取8 ml扩增后产物,进行琼脂糖电泳检测。
1.2.8 pET-28a-GFP重组质粒转化到表达菌BL-21
1) 取100 μl感受态
BL-21,冰上慢速解冻,均匀悬浮。加入2 μl经过酶切鉴定成功提取重组质粒pET-28a-GFP,轻轻混匀,冰上静置10-30 min。
2) 42℃水浴热激70 s,冰上放置2 min。
3) 加入200 μl 含抗生素的LB液体培养基,37 ℃,60 r/min震荡培养30 min。四支EP管中加入0.5 μl 的100 mM的IPTG诱导,另外EP管中不加入IPTG诱导。
4) 吸取200 μl菌液涂布于含抗生素的LB平板上,用涂布器均匀涂布,平皿正放静置1-2 h后,封口膜封好平皿, 37 ℃培养倒置12-16 h。
1.2.9 重组绿色荧光蛋白(GFP)的诱导表达
1) 取GFP重组菌接种于2 ml LB培养液(含Kana 100 mg/l)中,37 ℃ 150-220 r/min过夜培养。
2) 将20 μl菌液按1:50—100接种于2 ml LB培养液(含Kana 100 mg/l)中,37 ℃ 200r/min培养2h。
3) 按IPTG: LB培养基按1:1000加入2 μl的IPTG诱导表达,继续培养,对照组不加IPTG诱导。
4) 用紫外线照射菌体沉淀,保存照片。
2 结果与分析
2.1 提取质粒




图1 提取质粒时加入各溶液的现象
A. 加入溶液1浑浊;B. 加入溶液2变澄清;C. 加入溶液3出现白色絮状沉淀;D. 加入氯仿抽提溶液分层
2.2 重组质粒构建的鉴定
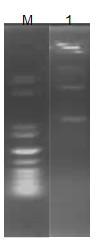
图2 酶切重组质粒pET-28a-GFP
Fig.2 indentification of pET-28a-GFP plasmid cleavage
Lane1:Marker protein;Lane 2:含目的基因的重组质粒,得到有两条带,可知前面**条带为GFP基因片段,第二条带为质粒载体,故成功构建了重组质粒pET-28a-GFP。
2.3 PCR检测结果
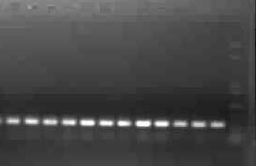
#p#分页标题#e#
图3 重组质粒的PCR鉴定
Fig. 3 PCR indentification of pET-28a-GFP
PCR扩增后产物经过电泳检测,可以看到明显的一条亮带,其大小约为750 bp。可知eGFP基因成功连接到pET-28a载体。其前面不明显的弥散的条带为引物二聚体。
2.4 IPTG诱导表达

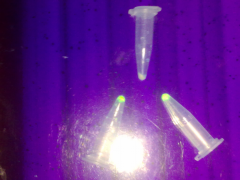
图 4 日光灯和紫外灯下的表达结果
在日光灯和紫外光下,携带重组质粒pET-28a-GFP的大肠杆菌
BL-21用IPTG诱导表达后呈现绿色,说明含外源基因GFP的质粒在大肠杆菌
BL-21体内成功表达;而没有经IPTG诱导的菌体则不发出绿色荧光。
3 讨论
本实验是一个综合性实验,与我们平时的实验相比对我们的要求更多更严。它主要要求我们形成认真思考的习惯,自己查阅本实验的相关文献,分析本实验的目的,通过本实验要达到什么样的结果,达到预计的结果需要通过怎样的方案实现,根据实验需要和自己的相关了解自己设置适合的实验方案,增强我们的思维能力和动手能力,同时也使我们的团队合作精神得到了提高,并学会把我们所学的知识有机的结合到一起。跟我们平时的实验相比较我们的实验素养可以得到很大的提高,有利于我们今后的学习和研究。通过本次实验我深刻的认识到了,在我们做研究的过程中一定要认真,每一步都不能出错,否则将功亏一篑,从头开始。
实验中重组质粒转化进入
E.coli DH 5α中的转化率很低,本组从
E.coli DH 5α中提取的质粒共9管,酶切发现有重组质粒存在的只有1管,转化率不高。造成重组质粒转化率不高的原因可能有以下几点:
(1)生长时期:实验发现在对数中期的大肠杆菌易生感受态,转化时菌浓度应控制在不超过107个/ml。浓度过高或者过低都会影响转化效率。
(2)CaCl
2法0 ℃放置时间的影响:细菌经0 ℃ CaCl
2处理后转化率随时间的推移而增加,24 h达到**高,之后转化率逐渐下降。
(3)化合物及无机离子的影响:在钙离子的基础上,联合其他二价金属离子或还原剂等物质处理细菌,可使转化率提高100-1000倍。
(4)质粒大小、构型的影响:用于转化的质粒DNA应主要是超螺旋态DNA。转化效率与外源DNA的浓度在一定范围内成正比,但当加入外源DNA的量过多或体积过大时,转化效率就会降低。1 ng超螺旋DNA即可使50 ul的感受态细胞达到饱和。一般情况下DNA溶液的体积不应超过感受态细胞体积的5%。
(5)防止杂菌和杂DNA的污染:整个操作过程均应在冰浴低温和无菌条件下进行,所用器皿,如离心管、EP管等均应彻底洗净,并经高压灭菌处理,所有的试剂都要灭菌,且注意防止被其他试剂、DNA酶或杂DNA所污染,否则均会影响转化效率或杂 DNA的转入,为以后的筛选、鉴定带来不必要的麻烦。
(6)试剂的质量:所用的试剂,如CaCl
2等均需是**高纯度的,并用超纯水配制,**好分装保存于干燥的冷暗处。
(7)42 ℃热处理时间很关键,转移速度要快,且温度要准确,同时注意热处理过程中离心管不要摇动。
(8)菌液涂平皿操作时,应避免反复来回涂,因为感受态细胞的细胞壁有了变化,过多的机械挤压涂布会使细胞破裂,影响转化率。
在重组质粒转化
BL-21 细胞后,在平皿中用IPTG诱导长出菌苔,但未出现单菌落,可能的原因有:(1)菌液没有涂开;(2)涂完后未先正放1h使菌液充分被LB吸收;(3)平皿上有水珠,未除去;(4)摇菌时间过长,菌过多。**后经过IPTG诱导表达,本组中未有一管表达成功,仅有老师的两管表达出绿色的荧光蛋白,表达量很低,可能的原因:(1)在转化过程中,由于操作不当,使重组质粒本身转化进入
BL-21的量很少,故经IPTG诱导表达量就很少。(2)pET-28a质粒在菌体中的拷贝数本来就很低,和其他质粒相比量会少一些,可以多摇一段时间,16-18h都可以,略微提高抗生素的浓度,有助于提高拷贝数,加入IPTG后,有助于提高表达量。(3)由于表达菌株
BL-21没有end-A突变,质粒的完整性和保真性都不如
DH5α,因为裂解时会有大量多糖干扰,质和量都不如
DH5α,pET质粒拷贝数很低,故重组质粒转化
BL-21的效率相对较低;
DH5α是重组酶缺陷型,质粒在细胞内相对稳定,而
BL-21为非重组酶缺陷型,转化进入其中的重组质粒不能稳定存在,故GFP诱导表达量就相对较低。



